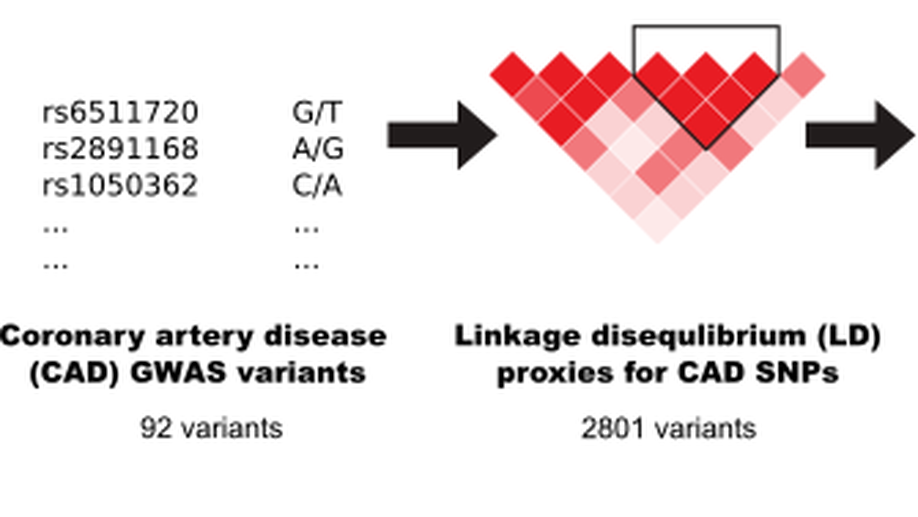
CRISPR perturbations at many coronary artery disease loci impair vascular endothelial cell functions
Genome-wide association studies have identified 161 genetic variants associated with coronary artery disease (CAD), but the causal genes and biological pathways remain unknown at most loci. Here, we used CRISPR knockout, inhibition and activation to target 1998 variants at 83 CAD loci to assess their effect on six vascular endothelial cell phenotypes (E-selectin, ICAM1, VCAM1, nitric oxide, reactive oxygen species, calcium signalling). We identified 42 significant variants located within 26 CAD loci. Detailed characterization of the RNA helicase DHX38 and CRISPR activation at the FURIN/FES, CCDC92/ZNF664 and CNNM2 loci revealed a strong effect on vascular endothelial cell senescence.
Integrative analysis of genomic variants reveals new associations of candidate haploinsufficient genes with congenital heart disease
Congenital Heart Disease (CHD) affects approximately 7-9 children per 1000 live births. Numerous genetic studies have established a role for rare genomic variants at the copy number variation (CNV) and single nucleotide variant level. In particular, the role of de novo mutations (DNM) has been highlighted in syndromic and non-syndromic CHD. To identify novel haploinsufficient CHD disease genes we performed an integrative analysis of CNVs and DNMs identified in probands with CHD including cases with sporadic thoracic aortic aneurysm (TAA). We assembled CNV data from 7,958 cases and 14,082 controls and performed a gene-wise analysis of the burden of rare genomic deletions in cases versus controls. In addition, we performed mutation rate testing for DNMs identified in 2,489 parent-offspring trios. Our combined analysis revealed 21 genes which were significantly affected by rare genomic deletions and/or constrained non-synonymous de novo mutations in probands. Fourteen of these genes have previously been associated with CHD while the remaining genes ( FEZ1, MYO16, ARID1B, NALCN, WAC, KDM5B and WHSC1 ) have only been associated in singletons and small cases series, or show new associations with CHD. In addition, a systems level analysis revealed shared contribution of CNV deletions and DNMs in CHD probands, affecting protein-protein interaction networks involved in Notch signaling pathway, heart morphogenesis, DNA repair and cilia/centrosome function. Taken together, this approach highlights the importance of re-analyzing existing datasets to strengthen disease association and identify novel disease genes. ### Competing Interest Statement The authors have declared no competing interest.
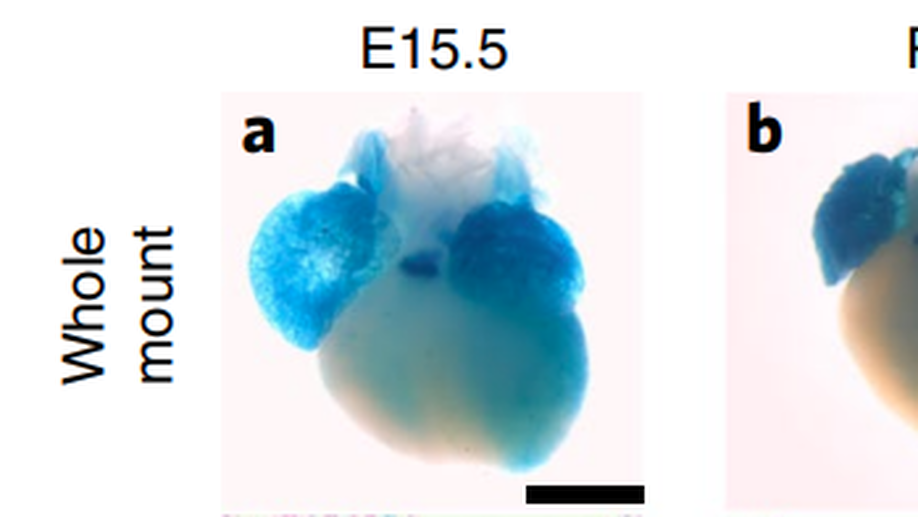
Loss of ADAMTS19 causes progressive non-syndromic heart valve disease
Valvular heart disease is observed in approximately 2% of the general population1. Although the initial observation is often localized (for example, to the aortic or mitral valve), disease manifestations are regularly observed in the other valves and patients frequently require surgery. Despite the high frequency of heart valve disease, only a handful of genes have so far been identified as the monogenic causes of disease2-7. Here we identify two consanguineous families, each with two affected family members presenting with progressive heart valve disease early in life. Whole-exome sequencing revealed homozygous, truncating nonsense alleles in ADAMTS19 in all four affected individuals. Homozygous knockout mice for Adamts19 show aortic valve dysfunction, recapitulating aspects of the human phenotype. Expression analysis using a lacZ reporter and single-cell RNA sequencing highlight Adamts19 as a novel marker for valvular interstitial cells; inference of gene regulatory networks in valvular interstitial cells positions Adamts19 in a highly discriminatory network driven by the transcription factor lymphoid enhancer-binding factor 1 downstream of the Wnt signaling pathway. Upregulation of endocardial Krüppel-like factor 2 in Adamts19 knockout mice precedes hemodynamic perturbation, showing that a tight balance in the Wnt-Adamts19-Klf2 axis is required for proper valve maturation and maintenance.
Copy number variation analysis in bicuspid aortic valve-related aortopathy identifies TBX20 as a contributing gene
Bicuspid aortic valve (BAV) is the most common congenital heart defect (CHD), affecting 1-2% of the population. BAV is associated with thoracic aortic aneurysms (TAAs). Deleterious copy number variations (CNVs) were found previously in up to 10% of CHD cases. This study aimed at unravelling the contribution of deleterious deletions or duplications in 95 unrelated BAV/TAA patients. Seven unique or rare CNVs were validated, harbouring protein-coding genes with a role in the cardiovascular system. Based on the presence of overlapping CNVs in patients with cardiovascular phenotypes in the DECIPHER database, the identification of similar CNVs in whole-exome sequencing data of 67 BAV/TAA patients and suggested topological domain involvement from Hi-C data, supportive evidence was obtained for two genes (DGCR6 and TBX20) of the seven initially validated CNVs. A rare variant burden analysis using next-generation sequencing data from 637 BAV/TAA patients was performed for these two candidate genes. This revealed a suggestive genetic role for TBX20 in BAV/TAA aetiology, further reinforced by segregation of a rare TBX20 variant with the phenotype within a BAV/TAA family. To conclude, our results do not confirm a significant contribution for deleterious CNVs in BAV/TAA as only one potentially pathogenic CNV (1.05%) was identified. We cannot exclude the possibility that BAV/TAA is occasionally attributed to causal CNVs though, or that certain CNVs act as genetic risk factors by creating a sensitised background for BAV/TAA. Finally, accumulative evidence for TBX20 involvement in BAV/TAA aetiology underlines the importance of this transcription factor in cardiovascular disease.
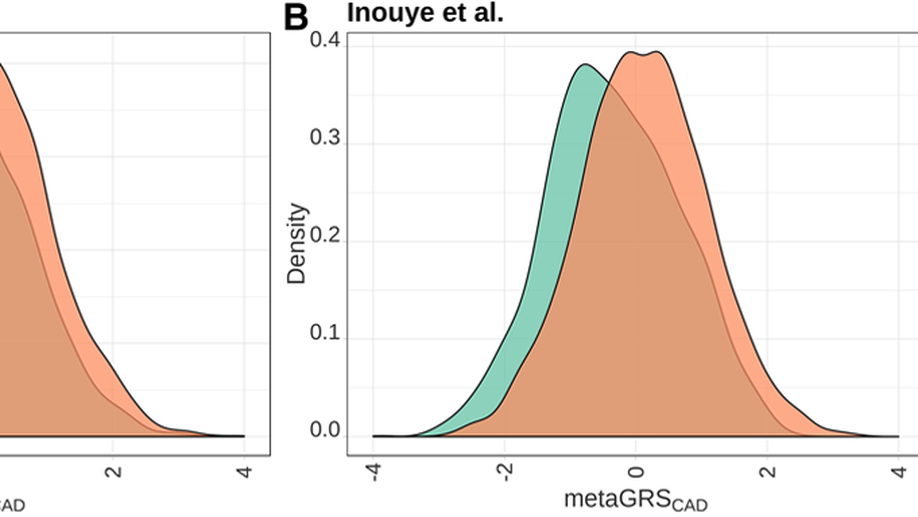
Validation of Genome-Wide Polygenic Risk Scores for Coronary Artery Disease in French Canadians
BACKGROUND: Coronary artery disease (CAD) represents one of the leading causes of morbidity and mortality worldwide. Given the healthcare risks and societal impacts associated with CAD, their clinical management would benefit from improved prevention and prediction tools. Polygenic risk scores (PRS) based on an individual’s genome sequence are emerging as potentially powerful biomarkers to predict the risk to develop CAD. Two recently derived genome-wide PRS have shown high specificity and sensitivity to identify CAD cases in European-ancestry participants from the UK Biobank. However, validation of the PRS predictive power and transferability in other populations is now required to support their clinical utility. METHODS: We calculated both PRS (GPSCAD and metaGRSCAD) in French-Canadian individuals from 3 cohorts totaling 3639 prevalent CAD cases and 7382 controls and tested their power to predict prevalent, incident, and recurrent CAD. We also estimated the impact of the founder French-Canadian familial hypercholesterolemia deletion ( LDLR delta >15 kb deletion) on CAD risk in one of these cohorts and used this estimate to calibrate the impact of the PRS. RESULTS: Our results confirm the ability of both PRS to predict prevalent CAD comparable to the original reports (area under the curve=0.72-0.89). Furthermore, the PRS identified about 6% to 7% of individuals at CAD risk similar to carriers of the LDLR delta >15 kb mutation, consistent with previous estimates. However, the PRS did not perform as well in predicting an incident or recurrent CAD (area under the curve=0.56-0.60), maybe because of confounding because 76% of the participants were on statin treatment. This result suggests that additional work is warranted to better understand how ascertainment biases and study design impact PRS for CAD. CONCLUSIONS: Collectively, our results confirm that novel, genome-wide PRS is able to predict CAD in French Canadians; with further improvements, this is likely to pave the way towards more targeted strategies to predict and prevent CAD-related adverse events.
ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm
Bicuspid aortic valve (BAV) is a common congenital heart defect (population incidence, 1-2%)1-3 that frequently presents with ascending aortic aneurysm (AscAA)4. BAV/AscAA shows autosomal dominant inheritance with incomplete penetrance and male predominance. Causative gene mutations (for example, NOTCH1, SMAD6) are known for $łeq$1% of nonsyndromic BAV cases with and without AscAA5-8, impeding mechanistic insight and development of therapeutic strategies. Here, we report the identification of variants in ROBO4 (which encodes a factor known to contribute to endothelial performance) that segregate with disease in two families. Targeted sequencing of ROBO4 showed enrichment for rare variants in BAV/AscAA probands compared with controls. Targeted silencing of ROBO4 or mutant ROBO4 expression in endothelial cell lines results in impaired barrier function and a synthetic repertoire suggestive of endothelial-to-mesenchymal transition. This is consistent with BAV/AscAA-associated findings in patients and in animal models deficient for ROBO4. These data identify a novel endothelial etiology for this common human disease phenotype.
At the Heart of a Complex Disease `Molecular Genetics of Congenital Heart Disease'
Congenital heart disease (CHD) is the most common type of birth defect and affects almost 1% of the general population. Compared to other rare congenital disorders, CHD rarely shows strictly Mendelian inheritance patterns. Human genetic studies have revealed that multiple genes contribute to the disease in pathways, which affect early cardiac development. Despite recent large-scale efforts to identify causal genes for CHD, the majority of cases remain enigmatic. The challenges in identifying genotype–phenotype relationships in CHD suggest a more complex pattern of inheritance, where structural as well as single nucleotide variants contribute to the disease and modifiers tune the spectrum of cardiac malformations expressed. Here, we review the current state of genetic research in CHD and discuss the challenges in moving variant identification in CHD into the personal genomics era. Key Concepts Key Concepts * Congenital heart disease (CHD) is a complex developmental phenotype with many genes contributing to its etiology. * Single nucleotide polymorphisms (SNPs) as well as structural variants contribute to the burden of CHD in the population. * Loss of function variants (LOF) and missense mutations can have different impacts during cardiac development, thus leading to different CHD phenotypes. * Genetic factors for congenital heart malformations can be inherited autosomal recessive, autosomal dominant, X-linked or show non-Mendelian patterns in families. * Genetic background can alter the manifestation of CHD and lead to different CHD subtypes or buffer against disease.
Family Based Whole Exome Sequencing Reveals the Multifaceted Role of Notch Signaling in Congenital Heart Disease
Left-ventricular outflow tract obstructions (LVOTO) encompass a wide spectrum of phenotypically heterogeneous heart malformations which frequently cluster in families. We performed family based whole-exome and targeted re-sequencing on 182 individuals from 51 families with multiple affected members. Central to our approach is the family unit which serves as a reference to identify causal genotype-phenotype correlations. Screening a multitude of 10 overlapping phenotypes revealed disease associated and co-segregating variants in 12 families. These rare or novel protein altering mutations cluster predominantly in genes (NOTCH1, ARHGAP31, MAML1, SMARCA4, JARID2, JAG1) along the Notch signaling cascade. This is in line with a significant enrichment (Wilcoxon, p< 0.05) of variants with a higher pathogenicity in the Notch signaling pathway in patients compared to controls. The significant enrichment of novel protein truncating and missense mutations in NOTCH1 highlights the allelic and phenotypic heterogeneity in our pediatric cohort. We identified novel co-segregating pathogenic mutations in NOTCH1 associated with left and right-sided cardiac malformations in three independent families with a total of 15 affected individuals. In summary, our results suggest that a small but highly pathogenic fraction of family specific mutations along the Notch cascade are a common cause of LVOTO.
Molecular Pathways and Animal Models of Hypoplastic Left Heart Syndrome
s Hypoplastic left heart syndrome (HLHS) is a rare and severe defect in which the structures of the left side of the heart are severely underdeveloped. Only a very small minority of HLHS cases can currently be explained. This review summarizes how growth of the left-sided structures of the heart is initiated very early during development and which mechanisms could be defective in HLHS. Numerous cascades driving development of ventricular cardiomyocytes have been described and are put into perspective. Current genetic, epigenetic, and hemodynamic concepts in HLHS pathogenesis are discussed in the context of both animal and human models of impaired growth of left-sided structures of the heart. Understanding the contribution of these factors may be crucial for stratification of therapeutic interventions in HLHS.
Aortic Dilatation Associated With a De Novo Mutation in the SOX18 Gene: Expanding the Clinical Spectrum of Hypotrichosis-Lymphedema-Telangiectasia Syndrome
Background: We report a 13-year-old female patient followed since birth for multiple rare congenital defects, including hypotrichosis, telangiectasia, and severe dilatation of the ascending aorta. Methods: Comprehensive phenotype assessment throughout childhood included repeated echocardiographic measurements, evaluation of renal function, and immunohistochemical analysis of skin biopsy samples. Whole-exome sequencing was performed for the patient and both unaffected parents. Results: We identified a novel de novo mutation in the transcription factor SOX18 (c.481C>T:p.Gln161*) in the patient, which was absent in all unaffected family members. Echocardiography revealed early onset and progressive dilatation of the ascending aorta. Skin biopsy results confirmed the defects of the blood vasculature in the presence of intact lymphatic vessels. Assessment of renal function did not show any signs of renal problems or renal failure in the patient. Conclusions: The genetic finding of a pathogenic SOX18 mutation enabled the diagnosis of the rare hypotrichosis-lymphedema-telangiectasia syndrome in our patient. The identification of a novel stop gain mutation in the SOX18 gene in association with dilatation of the aorta highlights the importance of this gene during the development of the circulatory system. Our study highlights the importance of whole-exome sequencing in the rapid identification of genes and gene mutations involved in rare conditions and thus expanding the knowledge and spectrum of clinical manifestations associated with them.

Mutations in SGOL1 cause a novel cohesinopathy affecting heart and gut rhythm
The pacemaking activity of specialized tissues in the heart and gut results in lifelong rhythmic contractions. Here we describe a new syndrome characterized by Chronic Atrial and Intestinal Dysrhythmia, termed CAID syndrome, in 16 French Canadians and 1 Swede. We show that a single shared homozygous founder mutation in SGOL1, a component of the cohesin complex, causes CAID syndrome. Cultured dermal fibroblasts from affected individuals showed accelerated cell cycle progression, a higher rate of senescence and enhanced activation of TGF-$β$ signaling. Karyotypes showed the typical railroad appearance of a centromeric cohesion defect. Tissues derived from affected individuals displayed pathological changes in both the enteric nervous system and smooth muscle. Morpholino-induced knockdown of sgol1 in zebrafish recapitulated the abnormalities seen in humans with CAID syndrome. Our findings identify CAID syndrome as a novel generalized dysrhythmia, suggesting a new role for SGOL1 and the cohesin complex in mediating the integrity of human cardiac and gut rhythm.